- Study protocol
- Open access
- Published:
Supplemental parenteral nutrition in critically ill patients: a study protocol for a phase II randomised controlled trial
Trials volume 16, Article number: 587 (2015)
Abstract
Background
Nutrition is one of the fundamentals of care provided to critically ill adults. The volume of enteral nutrition received, however, is often much less than prescribed due to multiple functional and process issues. To deliver the prescribed volume and correct the energy deficit associated with enteral nutrition alone, parenteral nutrition can be used in combination (termed “supplemental parenteral nutrition”), but benefits of this method have not been firmly established. A multi-centre, randomised, clinical trial is currently underway to determine if prescribed energy requirements can be provided to critically ill patients by using a supplemental parenteral nutrition strategy in the critically ill.
Methods/design
This prospective, multi-centre, randomised, stratified, parallel-group, controlled, phase II trial aims to determine whether a supplemental parenteral nutrition strategy will reliably and safely increase energy intake when compared to usual care. The study will be conducted for 100 critically ill adults with at least one organ system failure and evidence of insufficient enteral intake from six intensive care units in Australia and New Zealand. Enrolled patients will be allocated to either a supplemental parenteral nutrition strategy for 7 days post randomisation or to usual care with enteral nutrition. The primary outcome will be the average energy amount delivered from nutrition therapy over the first 7 days of the study period. Secondary outcomes include protein delivery for 7 days post randomisation; total energy and protein delivery, antibiotic use and organ failure rates (up to 28 days); duration of ventilation, length of intensive care unit and hospital stay. At both intensive care unit and hospital discharge strength and health-related quality of life assessments will be undertaken. Study participants will be followed up for health-related quality of life, resource utilisation and survival at 90 and 180 days post randomisation (unless death occurs first).
Discussion
This trial aims to determine if provision of a supplemental parenteral nutrition strategy to critically ill adults will increase energy intake compared to usual care in Australia and New Zealand. Trial outcomes will guide development of a subsequent larger randomised controlled trial.
Trial registration
NCT01847534 (First registered 5 February 2013, last updated 14 October 2015)
Background
In critical illness, enteral nutrition (EN) is usually delivered to provide estimated daily nutrition requirements via a gastric tube [1–3]. EN is the preferred choice of nutrition for critically ill adults because it mimics normal nutritional intake in health, preserves gastrointestinal tract (GIT) function, is relatively inexpensive and has been associated with a reduced incidence of pneumonia and mortality when started early after intensive care unit (ICU) admission [4–6]. The alternative to EN is parenteral nutrition (PN), which is a specialised solution designed to provide daily nutrition requirements intravenously. PN is used when a patient does not have a functioning GIT or when a clinical preference for use of PN exists [1, 7]. Until recently it was thought that PN was associated with an increased risk of infectious complications and mortality, although new data indicates that these risks may have reduced with contemporary care in the ICU [8, 9].
It has been reported that only 45–60 % [10] of energy is provided when EN is used alone due to delivery and tolerance problems [11], resulting in failure to meet daily energy requirements with unknown consequences. The strategy of “supplemental PN” aims to correct the energy deficit from inadequately delivered EN with a supply of PN, to meet 100 % of daily energy requirements in combination. This approach is based on the premise that delivery of close to 100 % of estimated daily nutrition requirements may improve patient outcomes. Whilst the strategy has been demonstrated to deliver close to 100 % of estimated energy needs, the effects on clinical outcomes have been contradictory [12–14]. A prospective randomised control trial (RCT) which investigated supplemental PN initiated early (within 48 hours of ICU admission) versus late (8 days after ICU admission) demonstrated that late supplemental PN resulted in patients being more likely to be discharged earlier from the ICU, with fewer infections when compared to patients in the early arm. However, late supplemental PN led to a higher proportion of hypoglycaemia, a more pronounced inflammatory response and did not affect overall hospital, 90-day mortality or functional status [15]. The outcomes from this study appear to be contradictory and may relate to the use of aggressive insulin therapy, which is not practiced in Australia and New Zealand (ANZ) [16]. Furthermore, the population in this RCT were largely patients undergoing cardiac surgery, and of low to moderate acuity. This patient group can usually return to volitional oral intake quickly and do not often require artificial nutrition due to their short duration of ICU stay; thus, it would seem there may be a low likelihood of benefit from supplemental PN in this population. Another RCT investigating supplemental PN from admission to ICU versus usual care found that the supplemental PN group received more energy (28 kcal/kg per day versus 20 kcal/kg per day) and had fewer nosocomial infections compared with the usual care group (27 % versus 38 %, respectively), but only on days 9–28 of ICU admission [14]. This finding may be explained by the positive effect of adequately delivered nutrition on immunity later in the ICU stay, which is also a biologically plausible explanation.
Thus, it seems that supplemental PN in addition to standard EN may be able to deliver increased energy to critically ill adults, but the exact clinical effects and the population that may benefit most remain undefined. Our aim is to determine if a supplemental PN strategy commenced 48–72 hours following ICU admission will deliver increased amounts of energy to adults with severe critical illness, when compared with usual care in six ANZ tertiary ICUs.
Methods
Design and study participants
A stratified, prospective, multi-centre, unblinded, randomised, parallel-group phase II study will be undertaken.
Inclusion criteria
-
1)
Admitted to intensive care between 48 hours and 72 hours previously
-
2)
Mechanically ventilated at the time of enrolment and expected to remain ventilated until the day after tomorrow
-
3)
At least 16 years of age
-
4)
Have central venous access suitable for PN solution administration
-
5)
Have one or more organ system failure related to their acute illness defined as:
-
a)
PaO2/FiO2 ≤ 300 mmHg
-
b)
Currently on one or more continuous vasopressor infusions which were started at least 4 hours ago at a minimum dose of:
-
Dopamine ≥ 5 mcg/kg/min
-
Noradrenaline ≥ 0.1 mcg/kg/min
-
Adrenaline ≥ 0.1 mcg/kg/min
-
Any dose of vasopressin
-
Milrinone > 0.25 mcg/kg/min)
-
6)
Renal dysfunction defined as
In patients without known renal disease:
-
a)
Serum creatinine > 171 mmol/L OR
-
b)
Currently receiving renal replacement therapy
In patients with known renal disease:
-
a)
An absolute increase of > 50 % in serum creatinine from baseline OR
-
b)
Currently receiving renal replacement therapy
-
7)
Currently has an intracranial pressure monitor or ventricular drain in situ
-
8)
Currently receiving extracorporeal membrane oxygenation
-
9)
Currently has a ventricular assist device.
Exclusion criteria
Patients will be excluded if:
-
1)
Both EN and PN cannot be delivered at enrolment (that is, either an enteral tube or a central venous catheter cannot be placed or clinicians feel that EN or PN cannot be safely administered due to any other reason)
-
2)
Currently receiving PN
-
3)
Standard PN solutions cannot be delivered at enrolment (that is, clinicians believe that a patient definitely needs a specific parenteral nutrition formulation (for example, glutamine supplementation or specific lipid formulation)
-
4)
Death is imminent or deemed highly likely in the next 96 hours
-
5)
There is a current treatment limitation in place or the patient is unlikely to survive to 6 months due to underlying illness
-
6)
More than 80 % of energy requirements have been satisfactorily delivered via the enteral route in the last 24 hours
-
7)
Are known to be pregnant
-
8)
The treating clinician does not believe the study to be in the best interest of the patient.
Randomisation, allocation concealment and blinding
Concealed randomisation will be performed via a web-based system which includes randomisation in blocks of 6 at each site. Treatment allocation will be stratified by site. The trial is unblinded.
Trial intervention and comparator
The intervention is the delivery of a supplemental PN strategy using Olimel N9-840E/Triomel 9, manufactured and supplied by Baxter Healthcare Corporation, Old Toongabbie NSW 2146, Australia. A multi-trace element solution (10 ml), multi-vitamin (Cernevit, Baxter Healthcare Corporation, 5 ml) and ascorbate (300 milligrams) for stability will be added to the intervention in a Baxter Healthcare Corporation compounding centre following good manufacturing practice.
Further details on the interventional product can be viewed at Additional file 1.
The comparator arm will be usual care, with provision and management of nutrition as per local practice at each participating site.
The intervention period is defined as 7 days from the day of randomisation.
Study procedures common to both arms
Patients will be screened for eligibility by research coordinators/medical staff at each site when they are between 48 and 72 hours of their first admission to the ICU. Those that are found to meet all the inclusion and none of the exclusion criteria will be randomised using a web-based randomisation system.
At randomisation, the body weight of study participants will be standardised using calculated body weight (CBW). To determine CBW, actual or estimated weight and height will be required to allow calculation of body mass index (BMI). The weight used to determine BMI will be defined according to the following hierarchy:
-
a)
Actual body weight if it has been recorded in the previous 6 weeks
-
b)
Estimated dry weight if actual weight is not known.
Height will be estimated using demi arm span [17].
CBW will be the patient’s actual weight if their BMI is deemed to be <25 kg/m2. If their BMI is ≥25 kg/m2, the CBW will be set to the patient’s ideal weight at a BMI of 23 kg/m2. Once the CBW has been determined, it will not be changed for the study duration.
Daily energy requirements will be estimated using CBW with a fixed prescription. The daily energy requirements will be set at 25 kcal/kg CBW unless the patient is receiving renal replacement therapy (RRT) and/or extracorporeal membrane therapy (ECMO), where 30 kcal/kg of CBW will be used. The daily energy requirement will only be changed during the study period if the patient commences or discontinues ECMO and/or RRT (with the two requirement options being 30 kcal/kg CBW or 25 kcal/kg CBW, respectively). A higher energy requirement has been chosen during RRT and/or ECMO due to the potential for increased metabolic stress and inflammation associated with the delivery of both therapies and the underlying disease processes that require these treatments [18]. Once randomised, the target rate for continuous EN delivery will be calculated by the treating clinical team to match the daily energy requirement, with the assumption that all patients should receive 100 % of their daily energy requirements from administration of EN and rounded up to the nearest 5 ml/hour. The choice of EN formula, protein requirement estimation and management of blood glucose levels will be according to local protocols.
Figure 1 demonstrates the study processes from screening to study completion.

Study overview. CRP: C-reactive protein; EN: enteral nutrition; EQ-5D: EuroQuol 5 dimension; ICU: intensive care unit; LFTs: liver function tests; LOS: length of stay; MV: mechanical ventilation; PN: parenteral nutrition; 6MWT: 6-minute walk test
Study procedures in the intervention arm
Day of randomisation:
-
The interventional product will be administered to intervention patients within 2 hours of randomisation via a central venous catheter (including long-term central catheters, for example, a Hickman catheter if already in situ) or a peripherally inserted central catheter. Management of the line will be as per the participating hospital’s usual procedure. Due to the increased risk of overfeeding with energy when PN is used, the intervention strategy has been designed to minimise this risk. Thus, the maximum amount of energy provided by the intervention will be 20 kcal/kg/day (or 24 kcal/kg/day for those on RRT and/or ECMO), which equals 80 % of the daily energy requirement set at 25 or 30 kcal/kg/day, respectively. This will allow for small amounts of energy provided by EN, 25/50 % glucose and propofol (non-nutritional energy sources) in addition to interventional product in the intervention arm.
The starting rate of PN will be determined by the amount of energy received via the enteral route in the 24 hours prior to randomisation:
-
a)
Between 40–80 % of daily energy requirement received from EN: PN rate will equal delivery of 10 kcal/kg of CBW/day (or 12 kcal/kg of CBW/day for those on RRT and/or ECMO)
-
b)
Less than 40 % of daily energy requirement received from EN: PN rate will equal delivery of 20 kcal/kg of CBW/day (or 24 kcal/kg of CBW/day for those on RRT and/or ECMO).
Management of EN in the intervention arm will be according to unit protocol. Every attempt will be made by the treating clinical team to achieve delivery of EN in the intervention arm to provide 100 % of daily energy requirements. Importantly, EN must not be reduced based on the amount of intervention being administered.
Daily review of intervention:
-
From study day 2 until study day 7 (or ICU discharge, whichever occurs first), the adequacy of energy from EN and non-nutritional sources will be assessed at midday by a member of the site research team. Total energy intake will be determined for the 24 hours prior to review and used to determine the rate of delivery of study PN for the subsequent 24 hours (Fig. 2). Once the rate is set for the following 24 hours by the research team, it should not be altered by the treating team unless deemed necessary for patient safety.
Fig. 2 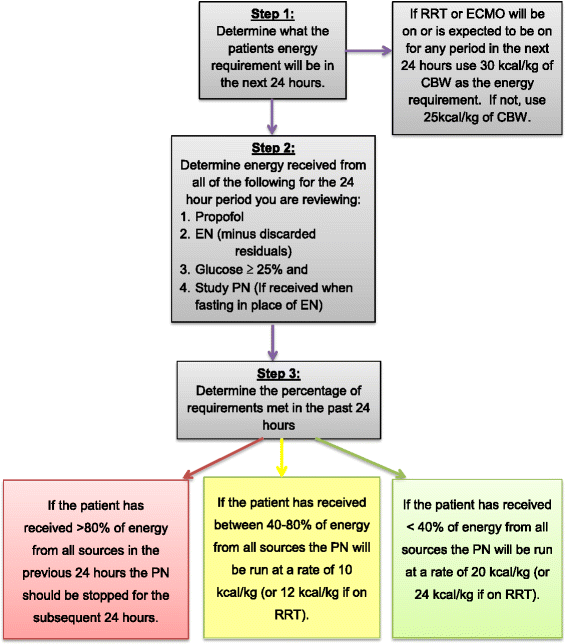
Daily adjustment of PN rate in intervention arm. ECMO: extracorporeal membrane oxygenation; EN: enteral nutrition; PN: parenteral nutrition; RRT: renal replacement therapy

Management of interruptions to EN in the intervention arm:
-
In the event of an anticipated or actual interruption to EN for a period of 2 hours or more in the intervention arm, the interventional strategy will be adjusted to minimise energy deficit for the period of the interruption. During the interruption period, the intervention will be run at the hourly rate corresponding to 20 kcal/kg or 24 kcal/kg for those on RRT and/or ECMO. If the patient is already receiving the highest rate of the intervention, there will be no change to the rate during the interruption period. As soon as is practical, EN should be recommenced as per local protocol and the intervention returned to the rate determined as per the midday assessment.
Cessation of study intervention prior to the end of the study period:
-
The intervention will cease either prior to ICU discharge or 7 days following enrolment if energy from EN and non-nutritional sources provides more than 80 % of estimated energy requirements on any day. Cessation on any one day will not preclude recommencement in the following 24 hours should the strategy be indicated based on the procedures previously outlined, until study day 7.
-
Should a patient commence oral intake during the 7-day study period, the intervention will cease when it is deemed that the patient will resume oral intake with the intent to provide nutrition, that is, not only to provide water or fluid intake.
Usual care arm
After enrolment, patients allocated to the usual care arm will commence or continue EN via an enteral tube to a target rate aimed to provide 100 % of daily energy requirements. All other aspects of nutrition therapy will be managed according to local unit protocol and, if required, include the use of promotility agents and the placement of nasojejunal feeding tubes prior to commencement of PN. PN will only be used in the usual care arm if the above methods have been attempted, or if an absolute contraindication to EN develops. The interventional product will be used in the usual care arm should PN be required within 7 days of randomisation. If PN is required after study day 7, it will be the usual hospital PN formula, managed by the treating clinicians as clinically appropriate.
Outcome measures
The primary outcome of this trial is the mean energy amount in calories delivered from nutrition therapy over the first 7 days of the study period.
Secondary outcomes include:
-
1)
Total protein amount delivered in the first 7 days of the study period
-
2)
Total energy amount delivered in the ICU stay (up to 28 days)
-
3)
Total protein amount delivered in the ICU stay (up to 28 days)
-
4)
Total antibiotic usage
-
5)
Sequential Organ Failure Assessment scores
-
6)
Duration of mechanical ventilation
-
7)
Duration of ICU and hospital stay
-
8)
Mortality to 180 days post randomisation
-
9)
Functional and quality of life to 180 days post randomisation
Study management and data collection
This trial will be coordinated by the Australian and New Zealand Intensive Care Research Centre (ANZIC-RC), Monash University, Melbourne, Australia. Dedicated study tools will be provided to participating sites to standardise all study procedures. Data will be collected at each site by dedicated and trained research staff using a paper case report form. Study variables collected will include baseline demographics such as anthropometric measurements, admission diagnoses, physiological parameters, Acute Physiology and Chromic Health Evaluation II, daily information including nutrition therapy, antibiotic use, blood tests and outcome data such as mortality, protocol deviations and serious adverse events (SAEs). At ICU and hospital discharge, functional, strength and health-related quality of life (HRQOL) assessments will be undertaken using the 6-minute walk test if possible and/or the highest level of function scale [19], hand grip strength and the EuroQol 5 dimension 5 level (EQ-5D-5 L) tools, respectively. Study participants will be contacted at 90 and 180 days post randomisation (unless previously deceased) to assess HRQOL, resource utilisation and survival. Follow-up assessments will be conducted via telephone by the research staff at the randomising site using a pre-prepared script to obtain the assessment using the EQ-5D-5 L. In the event that the patient is unable to complete the assessment at any time point, a relative or friend for the patient will be used as per the instructions for the EQ-5D-5 L. Data will be entered by the research staff at each participating site into a web-based database developed by Spiral Web Solutions, Wellington, New Zealand. Table 1 details the full table of events from baseline to outcome assessment.
Ethical considerations
The study protocol has been approved by The Alfred Hospital Ethics Committee in Australia and the Multi-Region Ethics Committee in New Zealand.
Participants in this trial will be unable to provide informed consent for themselves to participate in the study at the time of enrolment. A delayed consent model has been approved by the responsible ethics committees, which means a patient’s legal surrogate, relative/friend or whanau member will be approached for consent to participate in the study. Following consent from a patient’s legal surrogate, relative/friend or whanau member, the patient will be approached to give consent to continue in the trial if they recover the ability to do so and the timing is appropriate.
Sample size and power
Using two published RCTs on nutrition therapy in ANZ critically ill patients, we estimated that the usual care group would receive an average of 1,400 kcal/day. We aim to deliver an additional 420 kcal/day (using a standard deviation of 600 kcal/day) to the intervention group, which is a 30 % relative increase in energy delivery and requires a sample size of 100 patients (80 % power, significance 0.05).
This sample size will also provide baseline rates of other key secondary outcomes which could be used in the future to inform sample size estimations for larger RCTs assessing clinical outcomes.
Statistical analysis plan
Statisticians at the Australian and New Zealand Intensive Care Research Centre (ANZIC-RC) will perform statistical analysis using the intention-to-treat principle. All data will initially be assessed for normality and will be log-transformed as appropriate. Baseline variables and single measure outcomes will be compared using chi-square tests for equal proportion (or Fisher’s exact tests if numbers are small), Student’s t-test for normally distributed outcomes and Wilcoxon rank-sum tests otherwise. Continuously normally distributed repeated measure outcomes will be compared between groups using longitudinal mixed modelling fitting main effects for treatment and time with an interaction between treatment and time to determine if groups behave differently over time. Sensitivity analysis accounting for site, known covariates and baseline imbalances will also be performed for all outcomes, using logistic regression for binomial outcomes and mixed linear or non-linear modelling for continuous outcomes. Analysis will be performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA), and a two-sided p-value of 0.05 will be considered statistically significant.
Data and safety monitoring
Given the size of the trial, there are no planned interim analyses, and there is no dedicated data safety monitoring board. Safety will be monitored by reported adverse events and SAEs and reviewed by the study management committee (listed in Appendix 1) and Baxter Healthcare Corporation. All study sites will have an initial monitoring visit conducted by the project manager after two to five patients have been recruited. At this site visit, one intervention and one usual care arm patient will have 100 % source data verification at this visit; all other patients monitored at the visit will have consent procedures and eligibility criteria checked. Furthermore, intervention patients monitored at this initial visit will also have intervention delivery reviewed for adherence to the study protocol. Additional monitoring visits will be completed based on recruitment rates per site and any identified issues which need review after the initial monitoring visit.
The project manager will conduct remote monitoring of data completeness via the study website, and any data queries will be sent to the site for review.
Discussion
Nutrition is a commonly used therapy in the ICU. It is relatively inexpensive compared to other treatments and, if used correctly, may positively affect clinical and functional outcomes, although this remains to be definitively determined. Large-scale RCTs to date have failed to deliver EN to meet estimated energy requirements, or have delivered nutrition in a population or manner that makes the evidence difficult to translate into clinical practice. This study aims to determine if a supplemental PN strategy will safely deliver close to 100 % of energy requirements compared to usual care, identify a patient population who may benefit most and minimise the risks of overfeeding. This information will assist in the development of future studies to provide definitive answers on the role of energy intake in critical illness.
Trial status
The trial commenced recruitment on 17 February 2014. Final recruitment is expected to be achieved in late 2015 with 6 month outcomes available by early 2016.
Abbreviations
- ANZ:
-
Australia and New Zealand
- ANZIC-RC:
-
Australian and New Zealand Intensive Care Research Centre
- BMI:
-
body mass index
- CBW:
-
calculated body weight
- CI:
-
confidence interval
- ECMO:
-
extracorporeal membrane therapy
- EN:
-
enteral nutrition
- EQ-5D-5 L:
-
EuroQol 5 dimension 5 level
- FiO2 :
-
fraction of inspired oxygen
- GIT:
-
gastrointestinal tract
- HRQOL:
-
health-related quality of life
- ICU:
-
intensive care unit
- kcal:
-
kilocalorie
- kg:
-
kilogram
- mcg:
-
microgram
- min:
-
minute
- mmHg:
-
millimetre of mercury
- mmol:
-
millimole
- PaO2 :
-
partial pressure of oxygen
- PN:
-
parenteral nutrition
- RCT:
-
randomised controlled trial
- RRT:
-
renal replacement therapy
- SAE:
-
serious adverse event
References
Heyland DK, Dhaliwal R, Drover JW, Gramlich L, Dodek P. Canadian clinical practice guidelines for nutrition support in mechanically ventilated, critically ill adult patients. JPEN J Parenter Enteral Nutr. 2003;27(5):355–73.
Kreymann KG, Berger MM, Deutz NE, Hiesmayr M, Jolliet P, Kazandjiev G, et al. ESPEN Guidelines on Enteral Nutrition: Intensive care. Clin Nutr. 2006;25(2):210–23. doi:10.1016/j.clnu.2006.01.021.
Martindale RG, McClave SA, Vanek VW, McCarthy M, Roberts P, Taylor B, et al. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine and American Society for Parenteral and Enteral Nutrition: Executive Summary. Crit Care Med. 2009;37(5):1757–61.
Doig GS, Heighes PT, Simpson F, Sweetman EA, Davies AR. Early enteral nutrition, provided within 24 h of injury or intensive care unit admission, significantly reduces mortality in critically ill patients: a meta-analysis of randomised controlled trials. Intensive Care Med. 2009;35(12):2018–27.
Kompan L, Vidmar G, Spindler-Vesel A, Pecar J. Is early enteral nutrition a risk factor for gastric intolerance and pneumonia? Clin Nutr. 2004;23(4):527–32. doi:10.1016/j.clnu.2003.09.013.
Nguyen NQ, Fraser RJ, Bryant LK, Burgstad C, Chapman MJ, Bellon M, et al. The impact of delaying enteral feeding on gastric emptying, plasma cholecystokinin, and peptide YY concentrations in critically ill patients. Crit Care Med. 2008;36(5):1469–74. doi:10.1097/CCM.0b013e31816fc457.
Singer P, Berger MM, Van den Berghe G, Biolo G, Calder P, Forbes A, et al. ESPEN Guidelines on Parenteral Nutrition: intensive care. Clin Nutr. 2009;28(4):387–400. doi:10.1016/j.clnu.2009.04.024.
Doig GS, Simpson F, Sweetman EA, Finfer SR, Cooper DJ, Heighes PT, et al. Early parenteral nutrition in critically ill patients with short-term relative contraindications to early enteral nutrition: a randomized controlled trial. JAMA. 2013;309(20):2130–8. doi:10.1001/jama.2013.5124.
Harvey SE, Parrott F, Harrison DA, Bear DE, Segaran E, Beale R, et al. Trial of the route of early nutritional support in critically ill adults. N Engl J Med. 2014;371(18):1673–84. doi:10.1056/NEJMoa1409860.
Cahill NE, Dhaliwal R, Day AG, Jiang X, Heyland DK. Nutrition therapy in the critical care setting: what is "best achievable" practice? An international multicenter observational study. Crit Care Med. 2010;38(2):395–401. doi:10.1097/CCM.0b013e3181c0263d.
Passier RH, Davies AR, Ridley E, McClure J, Murphy D, Scheinkestel CD. Periprocedural cessation of nutrition in the intensive care unit: opportunities for improvement. Intensive Care Med. 2013. doi:10.1007/s00134-013-2934-8.
Bauer P, Charpentier C, Bouchet C, Nace L, Raffy F, Gaconnet N. Parenteral with enteral nutrition in the critically ill. Intensive Care Med. 2000;26(7):893–900.
Casaer MP, Mesotten D, Hermans G, Wouters PJ, Schetz M, Meyfroidt G, et al. Early versus late parenteral nutrition in critically ill adults. N Engl J Med. 2011;365(6):506–17. doi:10.1056/NEJMoa1102662.
Heidegger CP, Berger MM, Graf S, Zingg W, Darmon P, Costanza MC, et al. Optimisation of energy provision with supplemental parenteral nutrition in critically ill patients: a randomised controlled clinical trial. Lancet. 2013;381(9864):385–93. doi:10.1016/S0140-6736(12)61351-8.
Casaer MP, Mesotten D, Hermans G, Wouters PJ, Schetz M, Meyfroidt G et al. Early versus late parenteral nutrition in critically ill adults. N Engl J Med. 2011. doi:10.1056/NEJMoa1102662.
Finfer S, Chittock DR, Su SY, Blair D, Foster D, Dhingra V, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360(13):1283–97. doi:10.1056/NEJMoa0810625.
Estimating height in bedridden patients. RxKinetics, Plattsburg, MO 64477. http://www.rxkinetics.com/height_estimate.html. Accessed 9 June 2015.
Kagan I, Singer P. Nutritional imbalances during extracorporeal life support. World Rev Nutr Diet. 2013;105:154–9. doi:10.1159/000341292.
Hodgson C, Needham D, Haines K, Bailey M, Ward A, Harrold M, et al. Feasibility and inter-rater reliability of the ICU Mobility Scale. Heart Lung. 2014;43(1):19–24. doi:10.1016/j.hrtlng.2013.11.003.
Acknowledgements
This trial is funded by an unrestricted research grant by Baxter Healthcare Corporation. The study PN is provided at no charge for the study. Baxter Healthcare Corporation has not been involved in the original trial concept, development of the trial concept, trial management, data collection, analysis plans, interpretation of the data or preparation of the manuscript. Members of the Supplemental Parenteral Nutrition Clinical Investigators are as follows (listed as participating sites, principal investigators, research coordinators, dietitians and method centre staff):
Auckland City Hospital, Auckland New Zealand: Cardiothoracic and Vascular Intensive Care Unit; Shay McGuinness, Rachael Parke, Eileen Gilder, Lianne McCarthy, Keri-Anne Cowdrey, Rebecca Baskett
Auckland City Hospital, Auckland, New Zealand: Department of Critical Care Medicine; Colin McArthur, Lynette Newby, Lyn Gillanders, Varsha Asrani
Christchurch Hospital, Christchurch, New Zealand: Seton Henderson, Jan Mehrtens, Anna Morris, Emmeline Minto
University Hospital Geelong, Geelong, Australia: Neil Orford, Allison Bone, Tania Elderkin, Tania Salerno, Roy Hoevenaars
The Alfred, Melbourne, Australia: Owen Roodenburg, Meredith Young, Phoebe McCracken, Jasmin Board, Shirley Vallance, Emma Ridley, Eleanor Capel
Wellington Hospital, Wellington, New Zealand: Paul Young, Leanlove Navarra, Anna Hunt, Sally Hurford, Lynn Andrews, Diane Mackle, Catherine Boulton
Australian and New Zealand Intensive Care Research Centre: Michael Bailey, Andrew Davies, Adam Deane, Carol Hodgson, Emma Ridley.
The members of the management committee are listed in Appendix 1.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Competing interests
AD does sessional work with Baxter Healthcare Corporation independently of this trial.
SM has received speaking fees from Baxter Healthcare Corporation. Research in the Cardiothoracic and Vascular Intensive Care Unit, Auckland City Hospital is supported in part by an unrestricted grant from Fisher & Paykel Healthcare, New Zealand.
Authors’ contributions
ER contributed to the original trial concept, development of trial concept including statistical analysis plans, funding acquisition, drafting of the manuscript, trial management, is a management committee member, a site investigator and will assist with statistical analysis. AD contributed to the original trial concept, development of trial concept including statistical analysis plans, funding acquisition, drafting of the manuscript, trial management and is a management committee member. RP contributed to the development of trial concept including statistical analysis plans, drafting of the manuscript, trial management, is a management committee member and a senior site research coordinator. MB contributed to the development of the trial statistical analysis plan and will oversee analysis and assisted with drafting of the manuscript. CM contributed to the original trial concept, development of trial concept, drafting of the manuscript, trial management, is a management committee member and a principal investigator. LG contributed to the original trial concept, development of trial concept, drafting of the manuscript, trial management, is a management committee member and a site investigator. DJC contributed to the original trial concept, funding acquisition, is a management committee member and assisted with drafting of the manuscript. SM is the chief investigator, contributed to the development of trial concept including statistical analysis plans, drafting of the manuscript, trial management, is a management committee member and principal investigator. Members of the Supplemental Parenteral Nutrition Clinical Investigators contributed to trial management, study conduct at individual sites and drafting of the manuscript. All named and group authors read and approved the final version of the manuscript.
Additional file
Additional file 1:
Detailed product information for the interventional product. (DOCX 14 kb)
Appendix 1
Appendix 1
Management committee of the Supplemental Parenteral Nutrition in Critically Ill Patients Phase II Randomised Controlled Trial
Shay McGuinness, Emma Ridley, Andrew Davies, Rachael Parke, David (Jamie) Cooper, Lyn Gillanders, Colin McArthur, Neil Orford, Owen Roodenburg.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ridley, E.J., Davies, A.R., Parke, R. et al. Supplemental parenteral nutrition in critically ill patients: a study protocol for a phase II randomised controlled trial. Trials 16, 587 (2015). https://doi.org/10.1186/s13063-015-1118-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-015-1118-y